| Name | SimCH |
|---|---|
| Type | Single Cell and Spatial Omics |
| Version | v0.2 |
| Developers | Lei Sun, Gongming Wang, Zhihua Zhang |
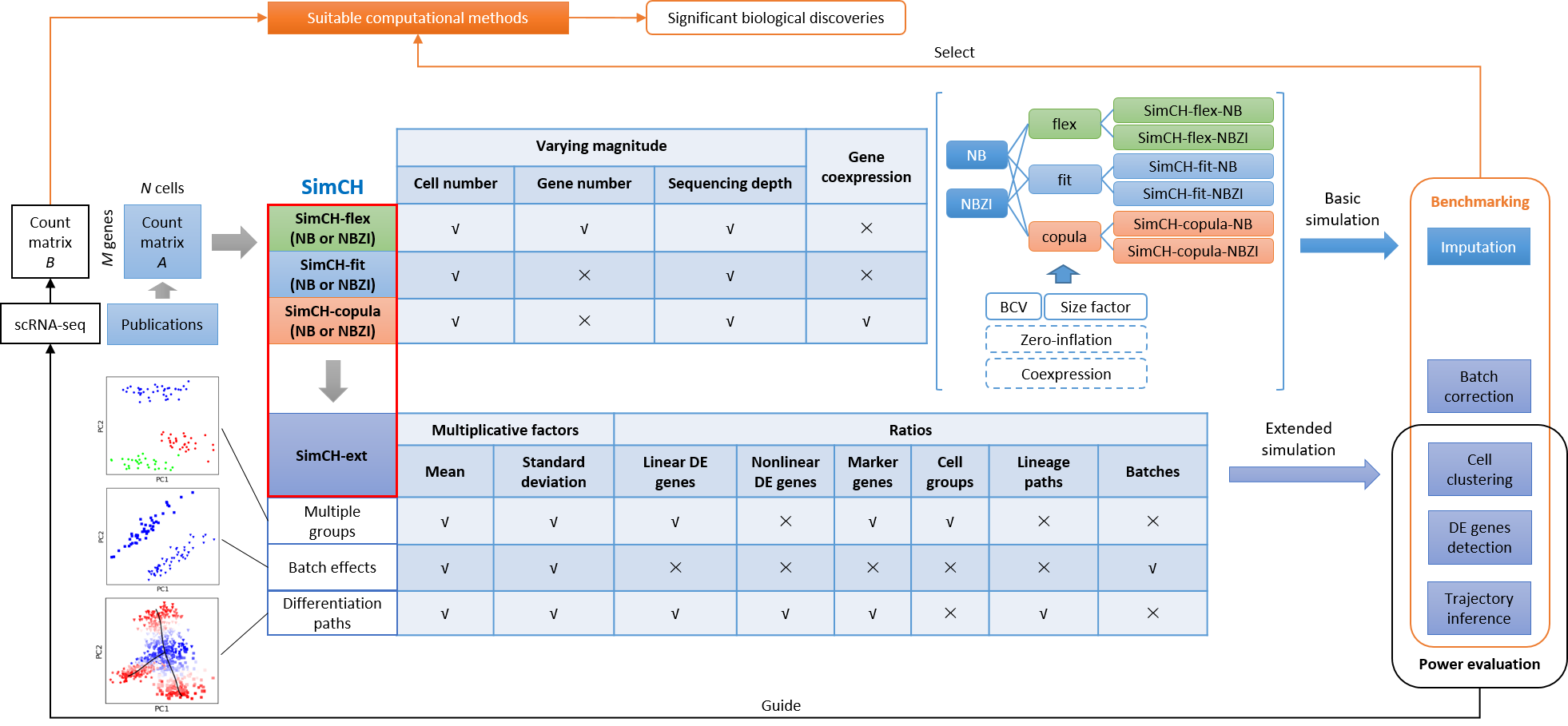
| Description | SimCH consists of three basic modes, i.e., SimCH-flex, SimCH-fit and SimCH-copula, as well as an extended mode, SimCH-ext, providing flexible magnitude configuration, good fit to experimental gene expression, gene coexpression preservation and complex simulation, respectively. Depending on the purpose of study, users may choose one of the three basic modes to estimate basic parameter settings from a homogeneous dataset. The SimCH-flex mode can generate simulated data with varying gene number, cell number and sequencing depth. The flexibility of SimCH-flex is achieved by building two logarithmic Gaussian mixture model (GMM) distributions to model gene mean expression and size factor respectively, and a scaled inverse chi-square distribution to model the biological coefficient of variation (BCV). The SimCH-fit mode can generate data to mimic the gene expression distribution of experimental data with varying cell number and sequencing depth. The SimCH-copula mode aims to retain the coexpression pattern amang genes in the experimental data by the Gaussian Copula framework. Importantly, a feature in SimCH modes allows users to set zero-inflation, or not, after NB modeling. This feature was inspired by the increasing number of studies showing that data sourced from UMI-based protocols (e.g., Droplet-based sequencing) can be modeled very well using NB distribution without zero-inflation. The SimCH-ext mode is designed to perform complex simulation for benchmarking the computational tools of cell clustering, DE gene detection, batch correction, and TI. Users can reset several parameters of SimCH-ext to mimic multiple groups, batch effects and differentiation paths, respectively. For the multi-group simulation, it is assumed that multiple cell groups evolve from an ancestral cell group estimated from the experimental data with a proportion of DE genes having mean expression shift controlled by multiplicative factors (MF). To simulate batch effects, the mean expression of all genes in the same batch of cells is assumed to shift with the same variation. When simulating differentiation paths, DE genes are randomly classified into linear and nonlinear categories, which evolve along differentiation paths by linear and nonlinear styles, respectively. Meanwhile, basic simulation can optionally generate a synthetic 'true' count matrix without loss of gene expression (e.g., dropouts), as well as the count matrix with expression loss modeled by Poisson sampling, which provides a reference for users to benchmark imputation methods. Benchmarking results could guide users to select a suitable tool or pipeline to achieve significant biological discoveries. In addition, users can assess the performance of specific methods with varying magnitude (i.e., cell number and sequencing depth). Such power evaluation can guide users toward choosing suitable cell number and sequencing depth in scRNA-seq experimental design to capture biological signals more accurately. |
| Downlaod | https://github.com/SIRG-YZU/SimCH |
| Article | https://academic.oup.com/bib/article-lookup/doi/10.1093/bib/bbac590 |
| Figure |
|
1 Beichen West Road, Chaoyang District Beijing 100101, China | 86-10-84097216
© China National Center for Bioinformation 2025, 京ICP备 10050270号-13


