| 名称 | 单细胞RNA测序数据仿真软件(SimCH) |
|---|---|
| 类别 | 单细胞与空间组学 |
| 版本号 | v0.2 |
| 开发者 | 孙磊,王公铭,张治华 |
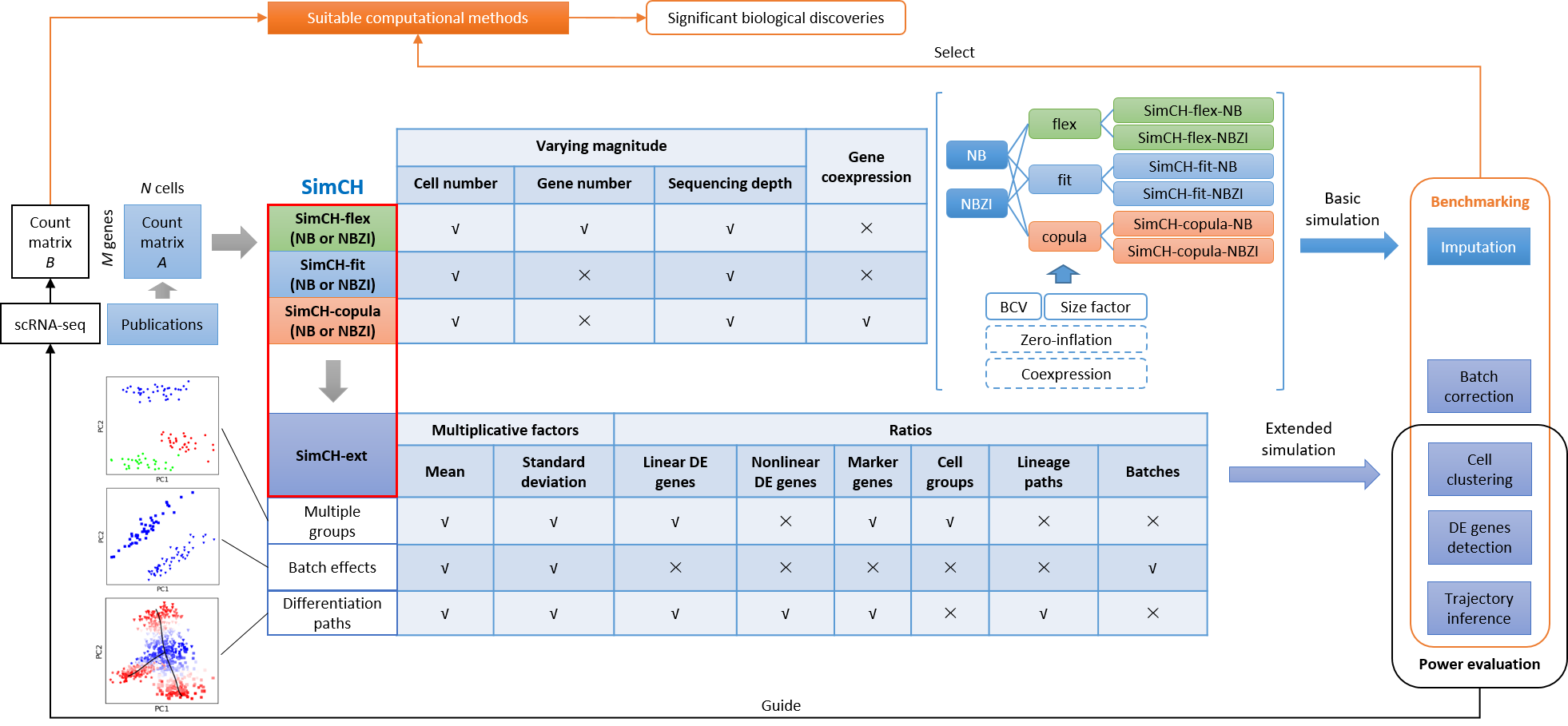
| 描述 | SimCH由三种基本模式组成,即SimCH-flex、SimCH-fit和SimCH-copula,以及扩展模式SimCH-ext,分别提供灵活的量级配置、与实验基因表达的良好拟合、基因共表达保存和复杂仿真。根据研究目的,用户可以从三种基本模式中选择一种,从同质数据集中估计基本参数设置。SimCH-flex模式可以生成具有不同基因数量、细胞数量和测序深度的仿真数据。SimCH-flex的灵活性是通过建立两个对数高斯混合模型(GMM)分布来实现的,这两个分布分别对基因平均表达和大小因子进行建模,并建立一个比例逆卡方分布来对生物变异系数(BCV)进行建模。SimCH拟合模式可以生成数据,以仿真具有不同细胞数量和测序深度的实验数据的基因表达分布。SimCH-copula模式旨在通过高斯copula框架在实验数据中保留基因的共表达模式。重要的是,SimCH模式中的一个功能允许用户在NB建模后设置零膨胀或不设置零膨胀。这一特征的灵感来自越来越多的研究,这些研究表明,来自基于UMI的协议(例如基于Droplet的测序)的数据可以在没有零膨胀的情况下使用NB分布很好地建模。SimCH-ext模式旨在执行复杂的仿真,以对细胞聚类、DE基因检测、批量校正和TI的计算工具进行基准测试。用户可以重置SimCH-ext的几个参数,分别仿真多组、批量效果和差异化路径。对于多组仿真,假设多个细胞组从根据实验数据估计的祖先细胞组进化而来,其中一定比例的DE基因具有由乘法因子(MF)控制的平均表达偏移。为了仿真批量效应,假设同一批细胞中所有基因的平均表达随着相同的变异而变化。在仿真分化路径时,DE基因被随机分为线性和非线性两类,它们分别以线性和非线性的方式沿着分化路径进化。同时,基本仿真可以选择性地生成没有基因表达损失(例如dropout)的合成“真实”计数矩阵,以及通过泊松抽样建模的表达损失的计数矩阵,这为用户基准插补方法提供了参考。基准测试结果可以指导用户选择合适的工具或管道,以实现重大的生物学发现。此外,用户可以评估不同程度的特定方法的性能(即细胞数量和测序深度)。这种能力评估可以指导用户在scRNA-seq实验设计中选择合适的细胞数量和测序深度,以更准确地捕获生物信号。 |
| 下载地址 | https://github.com/SIRG-YZU/SimCH |
| 文章发布 | https://academic.oup.com/bib/article-lookup/doi/10.1093/bib/bbac590 |
| 图片描述 |
|
北京市朝阳区北辰西路一号院104号 中国北京,100101 | 86-10-84097216
版权所有 © 国家生物信息中心 2025, 京ICP备 10050270号-13


